

Fenyloketonuria: Przyczyny, objawy i leczenie
Fenyloketonuria to jedna z bardziej niebezpiecznych chorób metabolicznych, która utrudnia życie. Przez wiele lat prowadziła do uszkodzeń mózgu i trwałego upośledzenia, dziś dzięki diagnostyce i szybkiemu wdrożeniu leczenia często pozwala na normalne funkcjonowanie. Konieczna jest jednak restrykcyjna dieta i przyjmowanie leków. Jak objawia się fenyloketonuria? Czy można ją zdiagnozować jeszcze przed narodzinami dziecka? Sprawdź, jakie są przyczyny tej groźnej choroby.

Składniki
Czym jest fenyloketonuria i jakie są jej przyczyny?

Fenyloketonuria (PKU) to choroba metaboliczna polegająca na zaburzeniach w przyswajaniu i rozkładaniu jednego z ważnych aminokwasów – fenyloalaniny. Schorzenie ma podłoże genetyczne i może być przekazane dziecku przez zdrowych rodziców, którzy są nosicielami zmutowanego genu. Za metabolizm fenyloalaniny odpowiada gen PAH – jego zadaniem jest rozkładanie tego aminokwasu na substancje niezbędne dla człowieka. W przeciwnym razie dochodzi do jego nagromadzenia się w organizmie i przeniknięcia do krwi w szkodliwej postaci. Zbyt wysokie stężenie fenyloalaniny trwale uszkadza mózg.
Jeśli mutacja genu PAH występuje tylko u jednego z rodziców, dziecko nie zachoruje, w przypadku dwojga rodziców z uszkodzonym genem, ryzyko wzrasta do 25% (i jest 50% szans na to, że stanie się nosicielem). Chorobę można wykryć już w ciąży, wykonując prenatalne badania DNA. Fenyloketonuria jest bardzo groźnym schorzeniem, brak szybkiej diagnostyki i leczenia prowadzi do trwałych uszkodzeń mózgu, a także niepełnosprawności umysłowej i fizycznej.
Składniki
Objawy fenyloketonurii
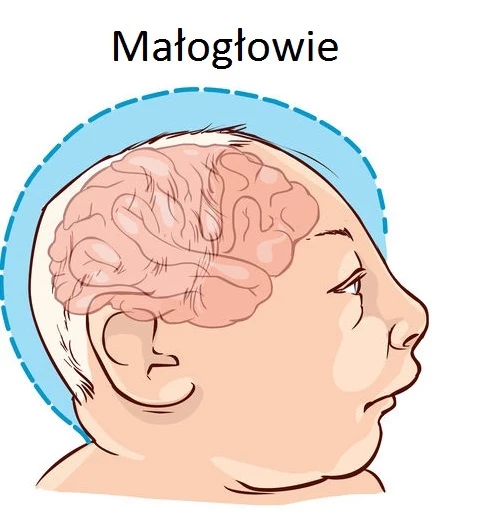
Pierwsze objawy choroby pojawiają się najczęściej w 3. miesiącu życia dziecka, rzadziej dopiero w 6. Niemowlę staje się apatyczne i traci zainteresowanie otoczeniem. Charakterystyczny jest też brak rozwoju psychoruchowego, bardzo słaby wzrost i przyrost masy ciała (a nawet jego brak), a także bardzo jasna skóra, pozbawiona pigmentu. U dzieci z fenyloketonurią często pojawiają się też wysypki na skórze, obniżone napięcie mięśniowe, drgawki i wymioty. Doświadczony lekarz jest w stanie zaobserwować też początki upośledzenia umysłowego. Inne symptomy tej choroby metabolicznej to mikrocefalia, czyli zbyt mała głowa, spłaszczona w czołowej i tylnej części, oraz nieprzyjemny, stęchły zapach skóry, moczu i wydobywający się z ust dziecka (niektórzy określają go jako "mysi" zapach). Bez leczenia objawy się nasilają, a w kolejnym etapie dochodzi do zachowań charakterystycznych dla autyzmu, problemów ze snem i mową, a także agresją w stosunku do siebie i innych.
Składniki
Diagnostyka i leczenie fenyloketonurii

Standardowo noworodki są badane pod kątem fenyloketonurii już w 3. dobie życia, a następnie po dwóch tygodniach (badanie polega na określeniu stężenia fenyloalaniny we krwi). Dzięki temu można wdrożyć leczenie, które chroni przed wystąpieniem objawów choroby. Podstawą jest wykluczenie lub znaczne ograniczenie (w zależności od stężenia fenyloalaniny we krwi określa się jej dzienną dopuszczalną dawkę) żywności zawierającej fenyloalaninę. Zalicza się do niej wszystkie produkty białkowe, a więc mięso, ryby, mleko, sery, jaja, rośliny strączkowe, orzechy, a także niektóre zboża, warzywa i owoce. Zakazane są także produkty zawierające popularny słodzik – aspartam (znajduje się w nim fenyloalanina).
Noworodki i niemowlęta, u których stwierdzono fenyloketonurię, nie mogą być karmione mlekiem matki ani zwykłym mlekiem modyfikowanym. Konieczne jest stosowanie specjalnego mleka dla dzieci z tym schorzeniem. Restrykcyjną dietę wykluczającą fenyloalaninę należy stosować do czasu zakończenia intensywnego rozwoju mózgu, czyli do 13-14. roku życia. Później można ją nieco złagodzić, chociaż lekarze twierdzą, że dla dobra całego organizmu, fenyloalaninę powinno się znacznie ograniczać do końca życia. Chorzy muszą też przyjmować specjalny preparat PKU (żywność medyczna) dostarczający do organizmu białko pozbawione szkodliwego aminokwasu. Jest to niezbędne do prawidłowego wzrostu i rozwoju.







